La enfermedad de Kawasaki es una vasculitis sistémica de etiología desconocida que afecta principalmente a niños menores de 5 años. Se incluye dentro de las vasculitis que afectan a arterias de mediano y pequeño tamaño. Es actualmente la primera causa de cardiopatía adquirida en la infancia en los países desarrollados, lo que la convierte en una enfermedad de suma trascendencia en el momento actual.
La enfermedad fue descripta por primera vez en Japón, en el año 1967 por el Dr. Tomisaku Kawasaki. Es reseñable que, la definición de caso propuesta por el Dr. Tomisaku Kawasaki se haya mantenido prácticamente sin cambios. Afecta predominantemente a niños de entre 6 meses y 5 años. Su presentación en menores de seis meses o en edades más tardías es menos frecuente, pero hay descritos casos en adolescentes y adultos, así como en el periodo neonatal. Afecta más a los niños que a las niñas. Se trata de la principal causa de enfermedad cardíaca adquirida durante la infancia en los países desarrollados.
Aunque esta enfermedad se ha descrito en todo el mundo, el mayor número de casos se ha notificado en los países del noreste de Asia, como Japón, Corea y Taiwán. La incidencia actual de enfermedad de Kawasaki en Japón es de más de 300 por cada 100.000 niños < 5 años (la más alta en todo el mundo).
Hasta el momento la causa de la enfermedad permanece desconocida pero se cree que existe una activación del sistema inmunológico por un agente infeccioso (aún no determinado) en personas genéticamente susceptibles. Existe mucha controversia sobre los probables mecanismos de activación del sistema inmunitario en los pacientes con enfermedad de Kawasaki. Estudios recientes sugieren que el agente causal de la Enfermedad de Kawasaki podría ser un agente medioambiental transportado por vientos troposféricos, posiblemente una toxina fúngica.
La Enfermedad de Kawasaki es una emergencia médica.
La enfermedad de Kawasaki también conocida como Síndrome de Kawasaki o Síndrome de los ganglios linfáticos mucocutáneos, es una vasculitis aguda o inflamación de los vasos sanguíneos que aparece esencialmente durante la infancia, generalmente autolimitada, y que tiene propensión por afectar las arterias coronarias. La enfermedad de Kawasaki se caracteriza por la aparición secuencial de una constelación de características clínicas. Muchas de sus manifestaciones clínicas pueden verse en otras enfermedades febriles comunes de los niños. Es por esta razón que el diagnóstico de la enfermedad de Kawasaki a menudo se considera un desafío clínico. Aproximadamente el 15-25 % de los pacientes no tratados pueden desarrollar aneurismas de las arterias coronarias (AAC).
Como informa ASENKAWA, “como representantes de las familias afectadas somos conocedores de la variabilidad en el diagnóstico, tratamiento y, sobre todo, seguimiento de los pacientes diagnosticados. Los síntomas de la enfermedad son similares a los de muchas patologías comunes en la infancia, no existiendo aún una prueba específica para su diagnóstico.
Para Marianela Pintos, presidenta de ASENKAWA, “visibilizar la enfermedad de Kawasaki no solo salva vidas, sino que mejora los resultados a largo plazo para los/as niños/as y sus familias, y contribuye a una mejor comprensión de una condición que aún necesita mucho más estudio y apoyo. También es fundamental para un diagnóstico precoz, Al ser más conocida la enfermedad, los padres y cuidadores pueden aprender a identificar los síntomas, ya que el diagnóstico y tratamiento a tiempo pueden prevenir complicaciones graves, especialmente las que afectan al corazón.

Síntomas comunes de la enfermedad de Kawasaki. Fuente: CDC
Síntomas de la Enfermedad de Kawasaki
“Esta patología se manifiesta con fiebre alta (alrededor de 39ºC), conjuntivitis bilateral no purulenta, eritema (enrojecimiento) labial y oral, eritema y edema de manos y pies, erupción en la piel (inflamada y/o irritada) y ganglios latero-cervicales.”, según explica la Dra.
Los síntomas de la enfermedad de Kawasaki incluyen fiebre superior a 102,2 grados Fahrenheit (39 grados centígrados) durante cinco o más días. Piel roja e hinchada en las palmas de las manos y en las plantas de los pies. Es posible que los síntomas no se presenten al mismo tiempo. Algunos niños tienen fiebre alta durante cinco o más días, pero presentan menos de cuatro de los síntomas necesarios para el diagnóstico de la enfermedad de Kawasaki. Podrían tener lo que se llama enfermedad de Kawasaki incompleta. Los niños con la enfermedad de Kawasaki incompleta siguen corriendo el riesgo de sufrir daños en las arterias del corazón.
¿Qué es La Enfermedad de Kawasaki? - Dr. Arturo Zuñiga
En los niños, la enfermedad de Kawasaki puede presentar síntomas similares a los de una afección denominada síndrome inflamatorio multisistémico.
Cuándo Consultar al Médico
Si tu hijo tiene fiebre durante más de tres días, comunícate con su profesional de atención médica.
Causas y Factores de Riesgo
Nadie sabe qué causa la enfermedad de Kawasaki. Sin embargo, los expertos no creen que la enfermedad se transmita de persona a persona. Algunos piensan que la enfermedad de Kawasaki ocurre después de una infección bacteriana o viral, o que está relacionada con factores ambientales.
Los factores de riesgo incluyen:
- Edad
- Sexo
- Origen étnico
La enfermedad de Kawasaki tiende a darse en determinadas estaciones.
Complicaciones de la Enfermedad de Kawasaki
La enfermedad de Kawasaki es una de las principales causas de enfermedades cardíacas en niños que viven en países desarrollados. Cualquiera de estas complicaciones puede dañar el corazón. La inflamación de las arterias del corazón puede debilitarlas y provocar un bulto en la pared de la arteria, llamado aneurisma. Los aneurismas aumentan el riesgo de formación de coágulos sanguíneos.

Complicaciones cardíacas asociadas a la enfermedad de Kawasaki. Fuente: Mayo Clinic
Diagnóstico y Tratamiento
En opinión de la Dra. Collado, “es fundamental hacer una detección precoz y un diagnóstico diferencial con patología infecciosa. Ante la mínima sospecha se debe realizar una ecocardiografía que permite su detección precoz”. Asimismo, -añade- “el pronóstico de los pacientes depende de la extensión y la gravedad (desde una simple dilatación a aneurismas) de la afectación de las arterias coronarias en el momento del diagnóstico y el seguimiento, por ello es clave la identificación rápida y precoz que evite cualquier complicación”.
En cuanto al tratamiento, la especialista detalla que “en etapas iniciales la administración de ácido acetil salicílico a dosis altas es fundamental para controlar la fiebre y evitar complicaciones cardiacas.
Como concluye ASENKAWA, “entre un 25% y un 30% de los niños que no recibe tratamiento sufre las secuelas de esta enfermedad, que en muchos casos afecta a su salud para siempre o requieren de controles a largo plazo. La única forma de prevenirla es conociendo su causa para poder tener armas para luchar contra ella.
No obstante, puntualiza que “los/as niños/as con enfermedad de Kawasaki pueden llevar una vida normal, teniendo en cuenta que si se mantiene la dilatación coronaria habrá que estar siempre muy pendientes de la posible aparición de dolor torácico súbito.
Vasculitis IgA (VIgA/PSH)
Las vasculitis son un grupo heterogéneo de enfermedades que se caracterizan por la inflamación de la pared de los vasos sanguíneos. Las características clínicas dependerán del tamaño, tipo y localización de los vasos afectados. La incidencia estimada de las vasculitis pediátricas se sitúa en 50 casos por cada 100.000 niños por año. Debido a la naturaleza heterogénea de las vasculitis y al conocimiento limitado de sus causas, es difícil establecer subgrupos adecuados. En la Conferencia de Consenso de Chapel Hill de 2012, se actualizaron las definiciones de vasculitis utilizando el mejor conocimiento sobre la etiopatogenia y hallazgos clínicos de los diferentes tipos.
Las pruebas de imagen son útiles, sobre todo en las vasculitis de medianos y grandes vasos, siendo en la mayoría de casos, necesaria la biopsia de tejidos afectos. Debido a la afectación multisistémica y a la baja incidencia, el diagnóstico es con frecuencia difícil y consecuentemente tardío, lo cual suele asociarse a una importante morbi-mortalidad.
Las VPV asociadas a IC se caracterizan por el depósito de inmunoglobulinas y/o factores del complemento predominantemente en las paredes de los pequeños vasos. La VIgA/ PSH es la forma más frecuente en Pediatría, siendo el resto raras en este grupo de edad, afecta a pequeños vasos con depósitos inmunes de IgA1. La IgA es la inmunoglobulina más importante de la inmunidad de mucosa y su glicosilación es fundamental para el aclaramiento de las moléculas IgA1.
La VIgA/PSH puede aparecer en todos los grupos de edad, siendo más frecuente durante la infancia, ocurriendo el 50% de los casos en menores de 5 años y el 75-90%, en menores de 10 años. La incidencia oscila entre los 10 y 20 casos por cada 100.000 niños menores de 17 años, pudiendo alcanzar los 70,3 casos/100.000 en el grupo de edad comprendido entre los 4 y 7 años. La distribución según el sexo es similar, aunque con predominio en varones en algunas series (1,5-2:1). Los afroamericanos rara vez se afectan y es algo más frecuente en niños con ascendente asiático.
La patogénesis de la enfermedad continúa siendo desconocida, aunque se le supone una base genética sobre la que actúan factores ambientales desencadenantes. La mayoría de los casos de VIgA/PSH son esporádicos, sin embargo, se han descrito determinadas asociaciones intrafamiliares. Se postula la participación de los genes del complejo mayor de histocompatibilidad en la patogénesis de la enfermedad. En este sentido, se ha relacionado en varios trabajos, ciertos polimorfismos del HLA-DRB1 y HLA-B*41:02 con la susceptibilidad de presentar una VIgA/PSH y con su gravedad. El HLA B35 y DQA1 se han relacionado con el riesgo de padecer una nefritis en pacientes con VIgA/PSH. El impacto de la expresión de los genes del sistema renina-angiotensina en la presentación y desarrollo de una VIgA/PSH se ha estudiado en varios trabajos.
Igualmente, se han encontrado asociaciones entre la VIgA/PSH y los genes que codifican moléculas relacionadas con la inflamación (citoquinas y moléculas de adhesión)(4). La VIgA/PSH aparece hasta en el 7% de los pacientes con fiebre mediterránea familiar (FMF), enfermedad autoinflamatoria determinada genéticamente. Estudios procedentes de Israel y Turquía muestran un aumento significativo de la prevalencia de mutaciones que afectan al gen MEFV, causa de la FMF, en los niños con VIgA/PSH en comparación con la población general.
La glicosilación aberrante de la región bisagra de la IgA1 se ha descrito como factor de riesgo para desarrollar una VIgA/PSH y nefritis por IgA (IgAN). Existen 2 subclases de IgA, la IgA1 y la IgA2. La IgA1 es la subclase predominante (80-90% de la IgA sérica), contiene una región bisagra con múltiples lugares de glicosilación. Se ha documentado la existencia de niveles séricos elevados de IgA1, complejos inmunes que contienen IgA1 (de pequeño peso molecular), IgA- ANCA, IgA- FR en los pacientes con VIgA/PSH. En aquellos que, además, presentan nefritis (VIgA/PSHN), se detectan complejos inmunes circulantes IgA1-IgG de gran masa molecular.
Durante los últimos años, muchos estudios han implicado a diferentes citoquinas proinflamatorias en la patogénesis de la enfermedad (TNF-α, IL-1-β, IL-2, IL-6, IL-8, TGF-β y VEGF). Se piensa que la activación del complemento es un importante factor para el daño tisular en la VIgA/PSH. El aclaramiento defectuoso de IC que contienen IgA por el sistema de complemento, juega un papel en la patogenia de la nefritis por IgA y la VIgA/PSH. La circulación de IC y alteraciones en la hemostasia pueden provocar daño vascular en la VIgA/PSH.
En algunos estudios, se ha relacionado la gravedad de la enfermedad con los niveles séricos de dímeros-D, complejo trombina-antitrombina, fragmentos de protrombina y factor de Von Willebrand, indicando estos hallazgos, probablemente, una reacción local en el vaso sanguíneo inflamado, más que una activación sistémica de la coagulación e hiperfibrinolisis. Una disminución marcada de la actividad del factor XIII podría dar lugar a complicaciones severas, tales como hemorragia intracraneal o hemorragia pulmonar, presumiblemente debido a una degradación específica del factor XIII por las enzimas proteolíticas liberadas por células inflamatorias, con defecto local de la hemostasia.
Manifestaciones Clínicas de la VIgA/PSH
La VIgA/PSH es una vasculitis sistémica con afectación multiorgánica. La lesión cutánea característica es la púrpura palpable, pudiendo presentar el paciente desde petequias a grandes equimosis, precediéndose con frecuencia de un exantema maculopapular eritematoso o urticarial. Aparecen de forma simétrica en las zonas declives (miembros inferiores y nalgas) (Figs. 1 y 2), aunque también pueden encontrarse en los brazos, cara, orejas y espalda.
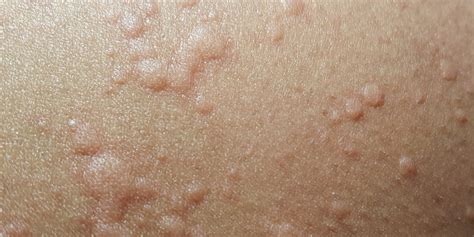
Manifestaciones cutáneas de la VIgA/PSH. Fuente: Semergen
Al inicio del cuadro, y sobre todo en niños pequeños, puede acompañarse de edema de cuero cabelludo, cara, manos, pies y escroto. Las lesiones ampollosas o hemorrágicas y necróticas son raras en los niños (2%), ocurriendo hasta en el 60% del paciente adulto.
Se describen en el 50-75% de los pacientes, siendo el primer síntoma de la enfermedad en el 14-36% de los casos. Se producen como consecuencia del edema y la hemorragia secundaria a la vasculitis de la pared intestinal, con mayor afectación del intestino proximal. El síntoma más frecuente es el dolor abdominal, generalmente se trata de un dolor cólico leve-moderado que puede acompañarse de vómitos; en algunos casos, el dolor puede ser muy intenso y limitante. Se encuentra sangre oculta en heces en el 56% de los pacientes, aunque la hemorragia intestinal masiva es rara (2%).
La invaginación es la complicación gastrointestinal más frecuente, limitándose al intestino delgado en el 60% de los casos, siendo la incidencia de esta complicación muy variable, hasta en un 2,3-3,5% en algunas series. La pancreatitis, el hidrops vesicular, la enteropatía pierde-proteína y la perforación intestinal son complicaciones raras, pero también descritas.
En un trabajo reciente, encuentran manifestaciones digestivas de la enfermedad en el 71% de los pacientes, sin tener ninguna manifestación cutánea el 7,6% de los mismos, llegándose al diagnóstico de VIgA/PSH tras endoscopia oral y biopsia(5). En este mismo trabajo, encuentran que en los pacientes con manifestaciones digestivas, los niveles de dímeros-D y de productos de degradación del fibrinógeno (PDF) están consistentemente más elevados que los marcadores inflamatorios (leucocitos, neutrófilos, VSG, PCR), pudiéndose utilizar estos como marcadores de afectación gastrointestinal en la fase aguda de la VIgA/PSH.
La artritis o artralgia puede ser el primer síntoma de la enfermedad en el 15-25% de los pacientes, encontrándose algún grado de afectación articular en el 82% de los mismos. Característicamente, la inflamación es poliarticular, dolorosa, sin eritema ni calor, pero con limitación, afectando con mayor frecuencia a las grandes articulaciones de miembros inferiores.
Un 30-50% de los pacientes con VIgA/PSH desarrollarán una glomerulonefritis (PSHN), pudiendo hacerse crónica y dar lugar a un daño renal permanente, motivo por el que es el factor pronóstico a largo plazo más importante de la enfermedad. Se manifestará con: hematuria microscópica/macroscópica, proteinuria, síndrome nefrótico/nefrítico, fracaso renal e hipertensión. En la mayor parte de los pacientes, la afectación es leve y autolimitada.
Los niños que no presentan alteraciones urinarias durante los 6 primeros meses de la enfermedad, no desarrollarán una disfunción renal en el seguimiento a largo plazo. El 20% de los pacientes con PSHN (7% de todos los casos de PSH) desarrollarán un síndrome nefrítico o nefrótico. La afectación renal se producirá durante las primeras 4 semanas de la enfermedad en el 75-80% de los pacientes, durante las primeras 6 semanas en el 91% y en los primeros 6 meses en el 97% de los casos, por lo que se aconseja realizar un seguimiento mediante uroanálisis durante, al menos, los seis primeros meses desde el inicio de la enfermedad.
Son raras, aunque la cefalea seguida de una ligera encefalopatía con mínimos cambios en el estado mental, tales como: labilidad emocional, apatía e hiperactividad, podría ser más frecuente de lo que se pensaba. Podemos encontrar alteraciones electroencefalográficas y convulsiones. La afectación pulmonar es rara.
Las manifestaciones escrotales y testiculares asociadas a la VIgA/PSH son relativamente comunes en los niños, en forma de: escroto agudo, epididimitis, orquitis y complicaciones del cordón espermático (hematoma y edema). Aunque la realización de una eco-doppler normalmente nos permite establecer un diagnóstico, puede llegar a ser necesaria la exploración quirúrgica para descartar la existencia de torsión testicular.
De forma poco frecuente, puede existir afectación ureteral durante la fase aguda de la enfermedad o tras resolución de la misma, en forma de obstrucción ureteral o ureteritis. La obstrucción puede ser uni o bilateral, parcial o total, secundaria a una vasculitis periureteral que puede desencadenar una isquemia ureteral.
Diagnóstico de la VIgA/PSH
El diagnóstico de la enfermedad es fundamentalmente clínico. No existen pruebas de laboratorio específicas para el diagnóstico de la enfermedad, por lo que nos basaremos fundamentalmente en los hallazgos clínicos, precisándose, en ocasiones, hallazgos anatomopatológicos.
En el estudio inicial, podremos encontrar anemia, leucocitosis con neutrofilia y un discreto aumento de la VSG y de la PCR; en algunos casos, una función renal y/o hepática alterada; en pacientes con proteinuria importante, podemos encontrar hipoalbuminemia. La trombocitosis se ha asociado con enfermedad más severa.
Los estudios estándares de coagulación son habitualmente normales, aunque la actividad del factor XIII se encuentra disminuida en relación con enfermedad más severa; no se aconseja su determinación rutinaria. La IgA se encuentra elevada en la mitad de los pacientes y no se correlaciona con la severidad de la enfermedad; pueden encontrarse IC circulantes de IgA. El significado de la presencia de IgA ANCA en la VIgA/PSH, está aún por dilucidar.
En general, el estudio básico inmunológico suele ser normal, encontrándose en alguna ocasión, niveles descendidos de C3 y C4. Podremos necesitar diferentes pruebas de imagen para conocer el alcance de la enfermedad gastrointestinal y urológica.
Tratamiento de la VIgA/PSH
El uso de prednisolona a 1-2 mg/kg (máximo, 60 mg) se podría considerar en niños con VIgA/PSH y dolor abdominal moderado-severo, una vez descartada patología abdominal potencialmente quirúrgica, como la invaginación y la perforación intestinal. En caso de vasculitis gastrointestinal muy severa (enteropatía pierde-proteínas y la hemorragia gastrointestinal severa), se ha descrito el éxito del tratamiento con: infusión de gammaglobulinas, pulsos de metilprednisolona, plasmaféresis e incluso bolos de ciclofosfamida.
El tratamiento de la VIgA/PSHN sigue siendo controvertido. La toma de decisiones terapéuticas es difícil dada la alta proporción de pacientes con pronóstico favorable y el curso clínico impredecible de pacientes individuales. En algunos estudios retrospectivos, el inicio tardío del tratamiento se asociaba a un peor pronóstico; por lo que, a pesar de la posibilidad de una remisión espontanea, podría ser aconsejable tratar a los pacientes severamente afectados tan pronto como sea posible.
Los efectos antihipertensivos y renoprotectores de los inhibidores de la enzima convertidora de la angiotensina (IECA) o de los antagonistas del receptor de la angiotensina II (ARA II) están bien documentados en el adulto con hipertensión y/o insuficiencia renal crónica. También, se ha constatado su eficacia en un estudio prospectivo realizado en niños con IgAN. Los corticoides orales o intravenosos forman parte de la mayoría de los regímenes terapéuticos y existe alguna evidencia sobre su efecto beneficioso a largo plazo en adultos con IgAN.
En un estudio reciente, se evaluaron de forma retrospectiva a 142 niños con VIgA/PSHN a los que se le realizó biopsia renal. Concluyen que el tratamiento precoz con IECA y ARA-II (tan pronto como la PSHN sea diagnosticada) se relaciona con la existencia de menor proteinuria, al menos, a medio plazo, y que el tratamiento precoz con pulsos de metilprednisolona parece disminuir el riesgo de secuelas(10). De forma análoga, a los pacientes con glomerulonefritis rápidamente progresivas de diferente etiología, la ciclofosfamida se ha utilizado en aquellos pacientes con VIgA/PSHN con manifestaciones más severas.
Otras terapias inmunosupresoras, tales como: azatioprina, micofenolato mofetilo, ciclosporina A o rituximab, han sido beneficiosas en casos individuales o pequeñas series de pacientes.
Pronóstico y Recurrencia
La VIgA/PSH es generalmente una enfermedad autolimitada (en 2-4 semanas), aunque hasta el 33% de los pacientes pueden presentar síntomas recurrentes (entre 1 y 6 episodios). Estas recurrencias suelen acontecer durante los 2-3 primeros meses, aunque se describen recaídas que sobrepasan los 18 meses del inicio de la enfermedad.
El pronóstico a largo plazo de los niños con VIgA/PSH se relaciona predominantemente con la existencia de enfermedad renal. Aunque ningún hallazgo es absolutamente predictivo, muchos estudios coinciden en que la presencia de síndrome nefrítico/nefrótico, la disminución de la actividad del factor XIII, la hipertensión, el desarrollo del fallo renal al inicio de la enfermedad y la presencia de esclerosis glomerular/semilunas/afectación tubulointersticial (lesiones histopatológicas clase IV y V), se considerarían como factores de mal pronóstico; de tal manera que, aunque puedan presentar una recuperación inicial, en el seguimiento a largo plazo (más de 20 años en algunos casos), casi la mitad de estos pacientes pueden presentar hipertensión o insuficiencia renal.
Aquellas mujeres a las que se les diagnosticó una VIgA/PSH durante la infancia, presentarán con mayor frecuencia hipertensión arterial y proteinuria durante el emb...
Enfoque Nutricional en la Enfermedad de Kawasaki
La nutrición desempeña un papel esencial en el proceso de recuperación y manejo de la enfermedad de Kawasaki.
- Evitar Alérgenos: En algunos casos, la enfermedad de Kawasaki puede desencadenar sensibilidades alimentarias.
- Hidratación Adecuada: Mantener una hidratación adecuada es crucial durante la enfermedad.
Enfocarse en una dieta equilibrada y adaptada a las necesidades individuales puede ser una herramienta poderosa en el camino hacia la recuperación.
tags: #kawasaki #enfermedad #enfoque #holistico