Las vasculitis se definen por la inflamación de la pared de los vasos sanguíneos, cuyo origen puede ser primario o secundario a otras patologías. El término vasculitis agrupa a un grupo heterogéneo de enfermedades.
La incidencia estimada de las vasculitis primarias pediátricas es aproximadamente de unos 50 casos/100.000 niños/año. Disponemos de criterios clasificatorios con el fin de poder agrupar grupos bien definidos, para poder realizar estudios y desarrollar protocolos. En 2006, se propusieron los criterios clasificatorios de consenso EULAR/PRES.
Actualmente, la anatomía patológica sigue considerándose el gold estándar para el diagnóstico de la mayoría de las vasculitis, con la excepción de la púrpura de Schönlein-Henoch y la enfermedad de Kawasaki, cuyo diagnóstico en la edad pediátrica es mayormente clínico.
Vasculitis por IgA (VIgA)
La vasculitis por IgA (VIgA), anteriormente conocida como púrpura de Schönlein-Henoch (PSH), es una vasculitis de pequeños vasos (vénulas, capilares y arteriolas) en los que se depositan inmunocomplejos IgA. Los órganos afectados con más frecuencia son piel, intestino y riñón, principalmente a nivel glomerular.
La vasculitis por IgA es la vasculitis pediátrica más frecuente. La VIgA supone la vasculitis sistémica más frecuente en edad pediátrica (90%). La incidencia oscila entre 3-26,7 casos por 100.000 niños/año.
La VIgA tiene un pico de presentación entre los 3 a 12 años, siendo raro que ocurra por debajo de los 2 años. El 50% de los casos ocurre en niños de 5 años o menores, y el 75-90% en menores de 10 años. Existe un discreto predominio en el sexo masculino (1,2:1 hasta 1,8:1 según series publicadas).
La mayoría de los casos son esporádicos, pero se han descrito asociaciones intrafamiliares y la susceptibilidad de padecerla en presencia de determinados polimorfismos de genes del complejo mayor de histocompatibilidad (HLA), incluyendo: HLA-DRB1 y HLA-B*4102. El HLA-B35 y DQA1 se han relacionado con el riesgo de padecer nefritis en pacientes con PSH.
La glicosilación aberrante de la región bisagra de la IgA1 predispone a la formación de inmunocomplejos y, por tanto, se ha descrito como factor de riesgo para desarrollar vasculitis por IgA y nefritis por IgA (IgAN). La activación del complemento en la VIgA es un importante factor de daño tisular, ya que el aclaramiento defectuoso de inmunocomplejos de IgA tiene un papel importante en la patogenia de la nefritis por IgA.
La VIgA es una vasculitis sistémica, por lo que puede afectar a cualquier órgano, siendo típica la afectación cutánea, articular, digestiva y renal. La sintomatología suele aparecer de forma progresiva en días o semanas, sin seguir un orden específico.
La afectación cutánea ocurre aproximadamente en el 75% de los niños con VIgA. La lesión más característica es la púrpura palpable en formas de petequias que pueden confluir, dando lugar a grandes equimosis. En ocasiones, puede presentarse como un exantema maculopapular eritematoso o urticarial. Las lesiones suelen aparecer en zonas declives (extremidades inferiores, glúteos), aunque también pueden afectarse las extremidades superiores, cara y tronco.
El inicio del cuadro clínico, sobre todo en los más pequeños, puede acompañarse de edema del cuero cabelludo, cara, manos, pies y escroto. Suelen aparecer en el 50-80% de los casos, principalmente en forma de artralgias de grandes articulaciones (tobillos y rodillas) y excepcionalmente como artritis. Suponen la primera manifestación de la enfermedad en el 15-25% de los pacientes.
Clínicamente, se manifiesta como inflamación periarticular sin eritema ni aumento de temperatura local, con dolor que limita la articulación afecta. Se describen en el 50-75% de los pacientes dentro de la primera semana de aparición de las lesiones cutáneas y dentro del primer mes de enfermedad, solo en el 11-20% suponen el primer síntoma.
La localización más frecuente es la zona proximal del intestino delgado, en forma de lesión isquémica, presentando sangre oculta en heces hasta en el 56% de los pacientes con afectación intestinal. En una publicación reciente se observó que el 71% de los pacientes presentaban manifestaciones digestivas; de ellos, el 7,6% sin afectación cutánea, llegándose al diagnóstico por biopsia vía endoscópica.
Un tercio de los pacientes presentará afectación renal, que suele manifestarse entre la 4ª y 6ª semana tras la aparición de las lesiones cutáneas. Se puede manifestar de diversas formas como: hematuria microscópica/macroscópica con o sin proteinuria, nefritis o síndrome nefrótico, hipertensión, insuficiencia renal, etc.
En la mayoría de las ocasiones, la afectación es leve y autolimitada, pudiendo ser grave en un 10%, aproximadamente; pudiendo cronificarse, dando lugar a un daño renal permanente. En el 97% de los casos, la afectación renal aparece en los 6 primeros meses. Las guías SHARE publicadas en 2019, para el diagnóstico y tratamiento de la VIgA, recomiendan realizar un control de la tensión arterial y sedimento urinario durante los primeros 6 meses.
Las manifestaciones aparecen, sobre todo en varones, en forma de: escroto agudo, epididimitis, orquitis y complicaciones del cordón espermático (hematoma y edema). La eco-doppler permite establecer un diagnóstico, aunque puede ser necesaria la exploración quirúrgica para descartar torsión testicular. La afectación pulmonar es rara.
El diagnóstico de la VIgA/PSH es clínico. A todo paciente con VIgA se le debe valorar la función renal al diagnóstico. El diagnóstico fundamentalmente es clínico. No existen pruebas complementarias específicas para llevar a cabo el diagnóstico, pero sí nos ayudan en el diagnóstico diferencial y en conocer el grado de afectación.
En el ámbito del laboratorio, en la fase aguda, podemos encontrar hallazgos inespecíficos (anemia, leucocitosis con neutrofilia…) y ligera elevación de VSG y PCR. El estudio de autoinmunidad (ANA, ANCA y FR) suele ser negativo, con niveles de complemento normales. En torno a un tercio de los casos pueden presentar elevación de IgA sérica, no siendo diagnóstica de la patología.
La biopsia cutánea será necesaria para el diagnóstico solo en aquellos casos con una presentación atípica. Desde el punto de vista histológico, el hallazgo típico es una reacción leucocitoclástica con depósito predominante de IgA en la pared de pequeños vasos, con una especificidad del 100%. La biopsia renal se lleva a cabo en casos de duda diagnóstica o con afectación renal grave, existiendo una buena correlación entre la gravedad y los hallazgos histopatológicos.
En la microscopía óptica, en la nefritis por IgA, se puede observar desde una proliferación mesangial leve hasta una glomerulonefritis con formación de semilunas grave. El diagnóstico diferencial se debe realizar, sobre todo en pacientes con presentación atípica.
El tratamiento principal es conservador. El uso de corticoides está indicado en afectación gastrointestinal severa, orquitis, vasculitis del sistema nervioso central, nefritis o hemorragia pulmonar. En función de las recomendaciones de tratamiento elaboradas por el grupo SHARE para la VIgA, el manejo de las artralgias/artritis y el dolor abdominal leve se puede realizar con analgesia convencional con paracetamol o AINES, evitando el uso de los últimos si existe compromiso renal.
En los pacientes con dolor abdominal intenso o sangrado rectal, afectación cutánea severa o artralgias intensas, se puede considerar el uso de corticoterapia, ya que ha demostrado reducir la intensidad y duración del dolor, además de prevenir la intervención quirúrgica. La dosis habitual es de 1-2 mg/kg/día de prednisolona (máximo 60 mg) durante 1-2 semanas en pauta descendente. En los pacientes con afectación abdominal y ante el posible síndrome de malabsorción secundario al edema de la pared intestinal, se recomienda su uso vía endovenosa.
En los casos con curso refractario, en los que se han utilizado corticoides con adecuada respuesta, se puede añadir un ahorrador de corticoides (colchicina, dapsona, micofenolato mofetilo, azatioprina, inmunoglobulinas, ciclosporina…). Las recomendaciones para el tratamiento de la nefritis por VIgA siguen siendo controvertidas ante la falta de evidencia científica. En la nefritis leve, el tratamiento de primera línea es prednisolona oral. En la nefritis moderada, el tratamiento de primera línea es prednisolona oral o metilprednisolona en pulsos endovenosos.
El pronóstico general es bueno. El pronóstico general es excelente, al ser una patología autolimitada a 2-4 semanas. Un tercio de los pacientes pueden presentar recurrencias de las lesiones cutáneas o dolor abdominal dentro de las primeras 6 semanas. A nivel renal, el pronóstico es bueno, con una resolución completa en cerca del 95% en aquellos pacientes que presentan hematuria y proteinuria.
Enfermedad de Kawasaki (EK)
La enfermedad de Kawasaki (EK) es una vasculitis sistémica aguda autolimitada, que afecta a vasos de pequeño y mediano calibre y puede llegar a producir complicaciones potencialmente graves. La EK es la segunda vasculitis más frecuente en la infancia tras la VIgA y supone la causa más común de enfermedad cardíaca adquirida en nuestro medio. La incidencia ha ido en aumento en los últimos años.
La incidencia en Europa se sitúa entorno los 5,4-15 casos/100.000 niños menores de 5 años/año. En un estudio realizado en Cataluña, la incidencia fue de 8 casos/100.000 niños menores de 5 años/año. En países asiáticos, la incidencia es mayor, siendo en Japón de 309 casos/100.000 niños menores de 5 años/año. El 77% de los casos ocurren en menores de 5 años, con un pico de incidencia entre los 18-24 meses, es poco frecuente en menores de 3 meses o mayores de 5 años; en estos grupos etarios, el riesgo de aneurisma coronario es mayor.
Hay un predomino en el sexo masculino de 1,5:1. El mayor pico de incidencia ocurre en primavera e invierno. La etiología es desconocida. Tras décadas de investigación aún no se conoce la etiología de la enfermedad. La teoría más aceptada es la existencia de un desencadenante infeccioso inhalado que infectaría las células epiteliales bronquiales ciliadas en personas genéticamente predispuestas.
Debido a la estacionalidad de la enfermedad, hay estudios en curso que orientan a un transporte del agente desencadenante por vientos de la troposfera. La alta incidencia en población asiática sugiere que la genética juega un papel importante en la enfermedad. Estudios del genoma humano han permitido identificar marcadores genéticos de susceptibilidad, severidad y refractariedad al tratamiento.
En alguno de estos estudios se han identificado genes del sistema HLA como: HLA B5, B44, Bw51, DR3 y DRB3*0301, que se asocian a enfermedad de Kawasaki en caucásicos. Además, se han identificado tres genes (ITPKC, ORAI1 y SLC8A1) relacionados con la vía de señalización del calcio, que activan la vía de la calcineurina y la traslocación del factor nuclear de células T activadas (NFAT), generando la transcripción de genes proinflamatorios (IL1B y TNF-alfa). Este hallazgo es importante, ya que tiene implicaciones terapéuticas, pudiendo utilizar la ciclosporina y el tacrólimus como tratamientos para inhibir la vía de la calcineurina.
El gen de la caspasa 3, en el cromosoma 4, se ha asociado a resistencia al tratamiento con inmunoglobulinas y afectación coronaria. En la EK participan tanto el sistema inmunológico innato como el adaptativo. La activación del sistema innato genera un gran número de neutrófilos circulantes, leucinas, IL1 e IL6, y factor de necrosis tumoral.
El inicio suele ser agudo, pudiendo estar precedido por síntomas respiratorios de vías altas o gastrointestinales. Las manifestaciones no aparecen secuencialmente, existiendo manifestaciones transitorias, de ahí la importancia de realizar una historia clínica completa. Los signos y síntomas clínicos en los pacientes no tratados desaparecen en una media de 12 días.
Síntomas Principales
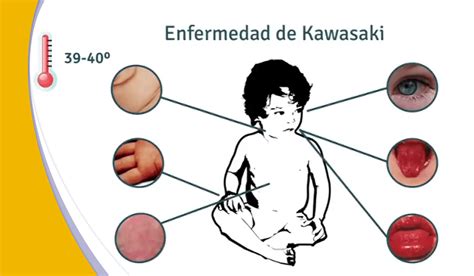
- Fiebre: Es el síntoma guía, suele ser persistente con escasa respuesta a antitérmicos, alcanzando picos de hasta 40ºC. La duración media sin tratamiento es de 1-3 semanas y en los tratados con inmunoglobulinas suele ceder a las 36 horas post-infusión.
- Conjuntivitis: Ocurre en el 85% de los pacientes y suele aparecer al inicio de la fiebre. Se manifiesta como inyección conjuntival bilateral, predominantemente bulbar no exudativa que resuelve sin secuelas.
- Labios y Boca: Los labios eritematosos, secos, fisurados con sangrado y la lengua aframbuesada son signos típicos. Además, podemos encontrar eritema orofaríngeo.
- Exantema: Se inicia durante los primeros 5 días desde el inicio de la fiebre. La erupción suele comenzar en el tronco, extendiéndose a extremidades, pudiendo confluir, sobre todo a nivel genital, donde produce una descamación temprana característica.
- Eritema y Edema: En la fase aguda es característico un eritema palmo-plantar y edema, en ocasiones, doloroso en dorso de manos y pies.
- Linfadenopatía: La linfadenopatía cervical anterior unilateral de más de 1,5 cm de diámetro no es la característica más típica, presentándose en menos del 50% de los casos.
La afectación puede ocurrir a cualquier nivel, manifestándose como: miocarditis, pericarditis, insuficiencia mitral (25%) o dilatación coronaria. En los primeros 10 días, no suelen detectarse los aneurismas, observándose inicialmente una hiperrefringencia de la pared en la ecocardiografía, apareciendo a partir de la 4-6ª semana. Algunos pacientes en la fase aguda pueden presentarse con shock cardiogénico (5%).
Otros manifestaciones son: artritis/artralgias de grandes articulaciones, dolor abdominal, vómitos y diarreas. A nivel respiratorio pueden presentar derrame pleural y, menos frecuentemente, infiltrado intersticial peribronquial. En lactantes es característica una marcada irritabilidad secundaria a meningitis aséptica. Otras manifestaciones neurológicas menos frecuentes son: sordera neurosensorial reversible, parálisis facial periférica unilateral y uveítis.
En el ámbito del laboratorio, hay marcadores que apoyan el diagnóstico como: elevación de reactantes de fase aguda (PCR, VSG, PCT, leucocitosis con neutrofilia, NT-proBNP); hiponatremia, hipoalbuminemia, transaminasitis y piuria estéril, así como la trombocitosis a partir del décimo día.
Se recomienda a todos los pacientes una valoración cardiológica con electrocardiograma y ecocardiograma. También debe realizarse un electrocardiograma, ya que es un método sensible, específico y seguro para la detección de dilataciones en las arterias coronarias y otras complicaciones cardíacas. El primer ecocardiograma deberá realizarse al momento del diagnóstico de la Enfermedad de Kawasaki y debe ser completo, con especial interés en la anatomía coronaria, la función ventricular, la función valvular y el pericardio.
Si el primer estudio fue normal, el segundo ecocardiograma deberá realizarse durante el transcurso de la segunda semana del inicio de los síntomas. A las 6/8 semanas si no se detectan alteraciones mediante ecocardiograma y las plaquetas han vuelto a la normalidad, se suspenderá la aspirina, en caso contrario se deberá seguir con el tratamiento con aspirina.
En los últimos dos años, a raíz de la pandemia por SARS-COV-2, el síndrome multisistémico inflamatorio (MIS-C), secundario a infección por COVID-19, se ha convertido en uno de los principales diagnósticos diferenciales, al compartir muchas de las características clínicas con la EK.
¿Qué es La Enfermedad de Kawasaki? - Dr. Arturo Zuñiga
Tabla comparativa de la incidencia de la enfermedad de Kawasaki en diferentes regiones:
| Región | Incidencia (casos/100.000 niños menores de 5 años/año) |
|---|---|
| Europa | 5,4 - 15 |
| Cataluña | 8 |
| Japón | 309 |
tags: #enfermedad #de #kawasaki #atipica