Las malformaciones congénitas pulmonares y de la vía aérea engloban un grupo heterogéneo de patologías que afectan al sistema respiratorio. Su origen se encuentra en un incorrecto desarrollo de diferentes estructuras pulmonares durante la embriogénesis. Nos encontramos ante una patología rara, con un aumento de casos en las últimas dos décadas debido al diagnóstico prenatal.
Para comprender las causas de estas malformaciones congénitas pulmonares, es preciso recordar la embriogénesis pulmonar. El desarrollo y maduración del aparato respiratorio comienza en torno a la cuarta semana de gestación y finaliza alrededor de los 8 años. Se describen cinco periodos evolutivos en la embriogénesis pulmonar:
- El periodo embrionario ocurre en las 6 primeras semanas de gestación. A partir del intestino primitivo, se origina un divertículo ventral, que dará origen al desarrollo del tejido epitelial del árbol respiratorio.
- El periodo pseudoglandular ocurre entre la 6ª-16ª semana de gestación; en este periodo se produce la ramificación dicotómica de los bronquios y termina con la formación de los bronquiolos terminales.
- Entre la semana de gestación 16 a 26 tiene lugar el periodo canalicular, donde tiene lugar la formación de los acinos y se inicia la irrigación de la vía respiratoria. Al final de esta etapa aparece el surfactante.
- Los bronquiolos terminales se transforman en bronquiolos respiratorios y se forman los sáculos, lugar en el que se realiza el intercambio gaseoso. Este periodo es conocido como periodo sacular, y tiene lugar entre la semana 28 a 36 de la gestación.
- El último periodo se denomina periodo alveolar; ocurre desde la semana 36 hasta los 8 años de vida. Durante este periodo, se forman los septos y las estructuras alveolares que seguirán su desarrollo posterior.

Figura 1. Fases del desarrollo embrionario pulmonar.
Clasificación de las Malformaciones Congénitas Pulmonares
Las malformaciones congénitas pulmonares se pueden clasificar en función de si predomina afectación parenquimatosa, vascular o mixta. Los defectos en el proceso de separación traqueal del intestino anterior y del desarrollo de las regiones ramificadas del pulmón son la base de la mayoría de las malformaciones pulmonares congénitas.
Agenesia Pulmonar
Supone la ausencia total o parcial de un pulmón con hiperplasia compensadora del contralateral. Son anomalías raras, con una incidencia de 1 por cada 15.000 recién nacidos. La agenesia se define, como la ausencia completa de tejido pulmonar, bronquio y arteria pulmonar (tipo I); que se diferencia de la aplasia pulmonar, porque esta presenta un bronquio rudimentario (tipo II). El pulmón contralateral es hiperplásico.
Este defecto congénito se puede presentar de forma aislada o asociado con otras anomalías a nivel esquelético, cardiovascular, gastrointestinal o genitourinario. Su pronóstico va a depender de las anomalías asociadas.
En la radiografía de tórax muestra ausencia de pulmón y desplazamiento del mediastino hacia el lado afecto, con disminución de espacios intercostales y elevación del diafragma. No existe un tratamiento curativo específico para esta patología. En la aplasia pulmonar, el esbozo bronquial puede dar lugar a infecciones de repetición, por lo que debe ser extirpado.
Hipoplasia Pulmonar
Podemos distinguir la hipoplasia pulmonar primaria de causa desconocida y la secundaria a otras patologías, siendo especialmente grave la asociada a hernia diafragmática congénita. Las manifestaciones clínicas de la forma primaria producen síntomas inmediatamente tras el nacimiento, manifestándose como distrés respiratorio grave. Además, la disminución de vascularización origina hipertensión pulmonar.
El manejo de la hipoplasia pulmonar primaria de los recién nacidos es conservador y depende de la gravedad de las alteraciones. En el caso de la hipoplasia pulmonar secundaria, se basa en la corrección quirúrgica de las malformaciones asociadas.
Hiperinsuflación Lobar Congénita
La hiperinsuflación lobar congénita es una patología poco frecuente, con una incidencia estimada de alrededor de 1 por cada 20.000 a 30.000 nacimientos. Atrapamiento aéreo distal a obstrucción bronquial por efecto valvular, con variable repercusión clínica y diagnóstico generalmente postnatal.
Se caracteriza por una alteración localizada del cartílago en la pared bronquial, ocasionando un efecto valvular y, consecuentemente, una hiperinsuflación y atrapamiento aéreo en uno o varios lóbulos pulmonares. El origen de la anomalía puede ser primario (ausencia de cartílago bronquial, estenosis bronquial o debilidad de la pared bronquial) o secundario a anomalías vasculares (sling de la arteria pulmonar o retorno venoso pulmonar anómalo). En el 50 % de los pacientes no se llega a identificar la causa.
En recién nacidos y lactantes puede manifestarse, como un distrés respiratorio grave, infecciones recurrentes, sibilancias, atelectasias debido a la compresión pulmonar o ser un hallazgo incidental. Dado que en el cribado prenatal la ecografía tiene baja sensibilidad, el diagnóstico suele realizarse de manera postnatal.
En la Rx tórax podemos encontrar distensión del lóbulo afecto con desplazamiento del mediastino con compresión y atelectasia del pulmón contralateral. Otras pruebas de imagen, como la RM o la TC, pueden definir la lesión y establecer la causa.
Figura 2. Área hipodensa bien delimitada en lóbulo medio con estructuras vasculares distribuidas de manera periférica.
Se considera tratamiento quirúrgico en pacientes sintomáticos, con lobectomía del lóbulo afecto.
Malformación Congénita de la Vía Aérea Pulmonar (MCVAP)
La malformación congénita de la vía aérea pulmonar (MCVAP), anteriormente conocida como malformación adenomatoidea quística, es la malformación pulmonar más frecuente, con una incidencia estimada de 1 caso por 8.500 a 35.000 recién nacidos. Grupo heterogéneo de malformaciones del árbol traqueobronquial con diferente expresión clínico-radiológica según la localización de la lesión.
El diagnóstico se realiza a través de la ecografía prenatal, en el segundo trimestre, entre la 21-24 semanas de edad gestacional, ofreciendo una imagen hiperecogénica, a veces difícil de diferenciar de otras malformaciones. Se caracterizan por un patrón anormal de las vías respiratorias, que tiene lugar durante la morfogénesis de la ramificación pulmonar. Estas malformaciones ocurren esporádicamente y no están asociadas a factores maternos, como edad, raza o exposiciones. En estudios recientes, se han identificado mutaciones asociadas a los genes FGFR2, DICER1, KRAS y TP53, lo que plantea un posible origen genético en determinados subtipos.
La presentación clínica de la MCVAP es variable, desde dificultad respiratoria grave en el periodo neonatal en relación con el tamaño de la lesión a pacientes asintomáticos. Según estudios recientes, alrededor del 25 % de los pacientes asintomáticos con diagnóstico de MCVAP desarrollan síntomas alrededor de los 6-7 meses de edad.
Los hallazgos en la Rx tórax son variables, dependiendo del tipo de MCVAP. Los tipos 1, 2 y 4 se caracterizan por quistes de paredes finas con ocupación interior o contenido aéreo, asociando una masa sólida en los tipos 1 y 4. El tipo 3 se presenta como una masa grande, sólida y homogénea. Además, en la Rx tórax podemos observar desplazamiento mediastínico hacia el lado contralateral, con hipoplasia del pulmón ipsilateral debido al efecto masa. Con esta prueba no es posible delimitar y definir bien las lesiones, por lo que nos apoyamos en técnicas de imagen avanzadas, como la TACAR, que nos permite determinar el tamaño de los quistes, la extensión anatómica e identificar otras alteraciones asociadas. Se debe realizar con contraste para evaluar la vascularización, especialmente si sospechamos asociación con secuestro pulmonar.
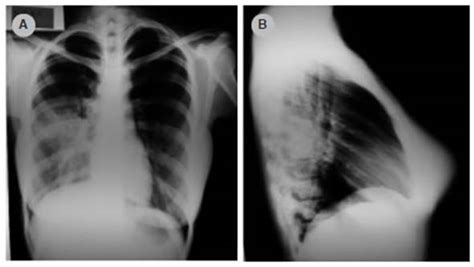
Figura 3. Lesión multiquística en lóbulo inferior izquierdo. Presenta quistes de pared fina, menores de 2 cm, con contenido aéreo en su interior.
Para el manejo prenatal de la MCVAP, es de gran utilidad la monitorización con ecografías seriadas del tamaño de la lesión en relación con el perímetro cefálico, mediante el cálculo de la ratio entre el volumen de la malformación y la circunferencia craneal (RVC). Si la RVC es superior a 1,6, se correlaciona con un alto riesgo de desarrollar hidrops fetalis y muerte fetal. En fetos de menos de 32 semanas con hidrops fetalis asociado a MCVAP estaría indicada, como terapia de primera línea, corticoides prenatales (betametasona). Aunque la respuesta es variable, han demostrado responder mejor los que presentan lesiones microquísticas.
La Rx tórax es una herramienta de primera línea, debido a su carácter no invasivo y rentabilidad. Se debería realizar antes del alta del recién nacido. Sin embargo, nunca debemos descartar una MCVAP prenatal basándonos en esta prueba. En la Rx tórax y TACAR postnatal se pueden observar uno o varios quistes o masa sólida con microquistes en su interior.
En los pacientes asintomáticos, con lesiones pequeñas y sin factores de riesgo de malignización, continúa la controversia sobre qué actitud es la más adecuada. Los expertos que apoyan la cirugía temprana, recomendada antes del primer año de vida, se basan en el riesgo de infección que dificultaría la cirugía posterior y en la posibilidad de malignización. El riesgo de malignización de las MCVAP se encuentra de manera general entre 1-3 %, siendo las de tipo 1 y 4 las que conllevan mayor riesgo, asociándose a carcinoma bronquioalveolar y blastoma pleuropulmonar tipo 1 (BPP), respectivamente.
Se han propuesto factores de alto riesgo para el desarrollo de BPP en niños con lesiones quísticas pulmonares asintomáticas. El síndrome DICER1 es un síndrome con patrón de herencia autosómico dominante con penetrancia disminuida, que predispone al cáncer hereditario y está relacionado con mutaciones en el gen DICER1 (presente en 2/3 de los casos de BPP).
Secuestro Pulmonar
Es una anomalía poco frecuente; supone el 1-6 %. Se clasifica en secuestro intralobar (SPI) y secuestro extralobar (SPE). El SPI se localiza dentro de un lóbulo pulmonar normal y carece de pleura visceral propia. Representa, aproximadamente, el 75 % de todos los secuestros pulmonares. El SPE se localiza fuera del pulmón normal y posee su propia pleura visceral.
La gran mayoría se localizan en hemitórax izquierdo, siendo la localización más frecuente entre el lóbulo inferior izquierdo y el hemidiafragma. En ocasiones, se localiza debajo del diafragma o en el retroperitoneo, en particular en la glándula suprarrenal, donde puede simular un neuroblastoma suprarrenal.
La presentación clínica es variable y depende del tipo, tamaño y localización de la lesión. Muchas de estas lesiones se diagnostican de forma prenatal. La mayoría de los recién nacidos son asintomáticos, pero, en ocasiones, presentan dificultad respiratoria en periodo neonatal. En el caso de SPI y las formas híbridas, se manifiestan en etapa de lactante y escolar, como neumonías de repetición, tos crónica, hemoptisis, etc., pero también como hallazgo casual en una Rx tórax.
Ante un diagnóstico de sospecha, la primera prueba a realizar es una Rx tórax, que muestra los secuestros pulmonares como una masa densa dentro de la cavidad torácica o parénquima pulmonar.

Figura 4. Imagen ilustrativa de un secuestro pulmonar.
La indicación de cirugía, lobectomía o resección segmentaria, se establece en aquellos secuestros pulmonares sintomáticos y en los SPI asintomáticos, por el riesgo de infección, o en las lesiones híbridas.
Atresia Bronquial
Se caracteriza por la interrupción intraútero de un bronquio lobar, segmentario o subsegmentario; como consecuencia, se produce una hiperinsuflación del segmento afecto e impactación mucosa, originando el mucocele o broncocele. Suele presentarse de manera incidental, al realizar una Rx tórax, como una zona hiperinsuflada o hiperlúcida que puede comprimir el tejido adyacente y provocar un desplazamiento del mediastino. El lóbulo más afectado es el superior izquierdo.
La mayoría de los pacientes se encuentran asintomáticos. La infección es poco frecuente dada la ausencia de comunicación con el árbol traqueobronquial.
Quiste Broncogénico
Es una lesión quística que se origina de un defecto en el desarrollo embrionario del árbol bronquial. Muchos pacientes con esta patología se encuentran asintomáticos y, con frecuencia, se descubren de manera incidental al realizar estudios por otros motivos.
En caso de ser sintomáticos, suelen presentarse, como tos recurrente, sibilancias, neumonías de repetición, dificultad respiratoria o disfagia. La TACAR suele ser la prueba diagnóstica de elección.
Diagnóstico Prenatal
El diagnóstico prenatal se realiza con la ecografía, que forma parte del cribado durante el embarazo. Es una prueba sensible para el diagnóstico y muy valiosa en el seguimiento y control evolutivo de estas malformaciones. La resonancia magnética fetal es una técnica de imagen que apoya a la ecografía en el abordaje de estas malformaciones.
Las principales herramientas en el diagnóstico postnatal son la radiografía de tórax (Rx tórax) y la angiografía por tomografía computarizada (angio-TAC). La radiografía de tórax es la prueba inicial de elección ante la sospecha prenatal de una malformación congénita pulmonar por su carácter no invasivo y su rentabilidad.
Tratamiento
Las manifestaciones clínicas son muy variables, dependiendo de la entidad y del grado de afectación, abarcando desde pacientes completamente asintomáticos hasta casos no compatibles con la vida. El tratamiento dependerá tanto de la repercusión clínica que implique cada malformación como de posibles riesgos futuros, individualizando cada caso en busca de un balance riesgo-beneficio óptimo. Hay casos que se manejan de manera expectante con diferentes tipos de controles sin llevar a cabo medidas terapéuticas excesivas y otros requerirán intervención urgente.