El término vasculitis se refiere a la inflamación y necrosis de los vasos sanguíneos, pudiendo afectar múltiples órganos o sistemas. Las manifestaciones clínicas varían dependiendo de la localización y tamaño de los vasos implicados. Estas pueden ser primarias o secundarias a infecciones, tumores, drogas u otras enfermedades reumáticas.
En niños, la frecuencia de vasculitis es baja, con excepción de la Púrpura de Schönlein-Henoch (PSH) y la enfermedad de Kawasaki (EK). Debido a la afectación multisistémica y la baja incidencia, el diagnóstico es con frecuencia difícil y tardío, lo cual suele asociarse a una importante morbi-mortalidad.
En 2005, la Liga Europea contra el Reumatismo (EULAR) y la Sociedad Europea de Reumatología Pediátrica (PRES) desarrollaron la primera clasificación pediátrica de vasculitis. El tratamiento generalmente se realiza con corticoides e inmunosupresores y se requiere un abordaje multidisciplinar.
¿Qué es La Enfermedad de Kawasaki? - Dr. Arturo Zuñiga
Púrpura de Schönlein-Henoch (PSH)
La púrpura de Shönlein-Henoch (PSH) es la vasculitis sistémica más frecuente en los niños. Sus manifestaciones clínicas más frecuentes son bien conocidas: púrpura palpable, artritis, dolor abdominal, sangrado intestinal y nefritis, aunque cualquier órgano puede verse afectado. Son consecuencia de una vasculitis leucocitoclástica de pequeños vasos debida al depósito de IgA1 en la pared de los vasos y del mesangio renal. Se ha asociado a una gran variedad de microorganismos, drogas y otros agentes ambientales.
La PSH puede aparecer en todos los grupos de edad, siendo más frecuente durante la infancia (entre los 3 y 15 años), ocurriendo el 50% de los casos en menores de 5 años y el 75-90%, en menores de 10 años. La incidencia oscila entre los 10 y 20 casos por cada 100.000 niños, pudiendo alcanzar los 70,3 casos/100.000 en el grupo de edad entre los 4 y 7 años. La distribución según el sexo es similar, aunque con predominio en varones en algunas series (1,5-2:1).
La patogénesis de la PSH continúa siendo desconocida; sin embargo, en general, se piensa que es una enfermedad mediada por el depósito de inmunocomplejos, caracterizados por la presencia de IgA1 polimérica, predominantemente a nivel de los capilares dérmicos, gastrointestinales y glomerulares.
Existen 2 subclases de IgA, la IgA1 y la IgA2. La IgA1 contiene una región bisagra con múltiples lugares de glicosilación. La glicosilación aberrante de esta región de la IgA1 podría explicar su participación exclusiva en la patogénesis de la PSH.
Se ha documentado la existencia de niveles séricos elevados de IgA1, complejos inmunes que contienen IgA1 (de pequeño peso molecular), IgA- ANCA, IgA- FR en los pacientes con PSH. En aquellos que, además, presentan nefritis (PSHN), se detectan complejos inmunes circulantes IgA1-IgG de gran masa molecular.
La lesión cutánea característica es la púrpura palpable, presentando el paciente desde petequias a grandes equimosis, pudiendo estar precedidas de un exantema maculopapular eritematoso o urticarial. Aparecen de forma simétrica en las zonas declive (miembros inferiores y nalgas), aunque también pueden encontrarse en los brazos, cara, orejas y espalda. Al inicio del cuadro, puede acompañarse de edema de cuero cabelludo, cara, manos, pies y escroto, sobre todo en niños pequeños.
Se describen en el 50-75% de los pacientes, siendo el primer síntoma de la enfermedad en el 14-36% de los casos. Se producen como consecuencia del edema y la hemorragia secundaria a la vasculitis de la pared intestinal. El síntoma más frecuente es el dolor abdominal de tipo cólico.
La artritis o artralgia puede ser el primer síntoma de la enfermedad en el 15-25% de los pacientes, encontrándose algún grado de afectación articular en el 82% de los casos. Característicamente, la inflamación es periarticular, dolorosa, sin eritema ni calor pero con limitación, afectando con mayor frecuencia a las grandes articulaciones de miembros inferiores.
Se producen en el 20-50% de los pacientes y es el factor pronóstico más importante de la enfermedad. La afectación renal se manifestará con: hematuria microscópica/macroscópica, proteinuria, síndrome nefrótico/nefrítico, fracaso renal e hipertensión, siendo la afectación severa en el 5-7% de los casos. Se desarrollará durante el 1º mes de la enfermedad en el 75-80% de los pacientes, y en el 97-100% de los casos a los 3 meses de inicio de la enfermedad.
Son raras, aunque la cefalea seguida de una ligera encefalopatía con mínimos cambios en el estado mental, tales como: labilidad emocional, apatía e hiperactividad, podría ser más frecuente de lo que se pensaba. Podemos encontrar alteraciones electroencefalográficas y convulsiones. La afectación pulmonar es rara. La orquitis es un hallazgo relativamente común en el niño con PSH, encontrándose hasta en el 24% de los casos.
El diagnóstico de la enfermedad es fundamentalmente clínico. No existen pruebas de laboratorio específicas para el diagnóstico de la enfermedad; por lo que, nos basaremos, fundamentalmente, en los hallazgos clínicos, precisándose en ocasiones hallazgos anatomopatológicos. La investigación irá encaminada a descartar otros posibles diagnósticos y a conocer la extensión de la afectación orgánica.
Si existe afectación renal, se recomienda realizar los controles y pruebas indicados en el Algoritmo 1. La biopsia cutánea, si se realizase (normalmente ante una presentación atípica y dudas diagnósticas), mostraría una vasculitis leucocitoclástica de pequeños vasos con depósitos de IgA e infiltración de neutrófilos y células mononucleares perivasculares. En la biopsia renal, podemos encontrar desde glomerulonefritis con lesiones focales y/o segmentarías hasta la formación de semilunas. También, encontraremos depósitos de IgA en la inmunofluorescencia.
El reposo disminuye la aparición de nuevas lesiones cutáneas, no precisando, en general, tratamiento específico añadido. En caso de lesiones bullosas, existen notificaciones del éxito del tratamiento con corticoides. El uso de prednisolona a 1-2 mg/kg (máximo, 60 mg) se podría considerar en niños con PSH y dolor abdominal moderado-severo, una vez descartada patología abdominal significativa, como la invaginación.
El tratamiento de la PSHN sigue siendo controvertido, no existiendo evidencia suficiente sobre la mejor guía de tratamiento para la PSHN establecida. Se proponen tratamientos con: prednisolona, metilprednisolona, ciclofosfamida, azatioprina, ciclosporina A, micofenolato mofetilo, dipiridamol, warfarina, plasmaféresis y rituximab.
La PSH es generalmente una enfermedad autolimitada (en 2-4 semanas), aunque hasta el 33% de los pacientes pueden presentar síntomas recurrentes (entre 1 y 6 episodios). Estas recurrencias suelen acontecer durante los 2-3 primeros meses, aunque se describen recaídas que sobrepasan los 18 meses del inicio de la enfermedad. Normalmente, los síntomas son similares a los del debut y parece ser que aquellos pacientes con afectación renal pueden recaer con mayor facilidad.
El pronóstico a largo plazo de los niños con PSH se relaciona predominantemente con la existencia de enfermedad renal. El tratamiento inicial de la PSH con corticoides no previene el desarrollo de nefritis.
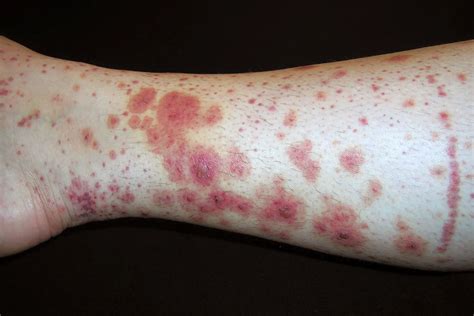
Púrpura de Schönlein-Henoch en un niño. Fuente: CUN
Enfermedad de Kawasaki (EK)
La enfermedad de Kawasaki (EK) es una vasculitis aguda y autolimitada. Su importancia se debe a que el 15-25% de los niños no tratados desarrollan anomalías coronarias (AC) que puede conducir a infarto de miocardio, muerte súbita o enfermedad isquémica cardiaca. Afecta a toda la edad pediátrica aunque, con mayor frecuencia, a menores de 5 años.
La incidencia es más elevada en países asiáticos. La EK afecta principalmente a niños entre 6 meses y 5 años, existiendo una mayor proporción de varones (1,5:1). La incidencia más alta se ha registrado en Japón. En nuestro país se estima que la incidencia anual acumulada es similar a la encontrada en Estados Unidos (20,8/100.000 niños menores de 5 años en 2006).
Las recurrencias son poco frecuentes (Japón 3% y Estados Unidos 1-2%). Los casos familiares son raros (0,7-2,1%) y el 50% aparecen en los 10 días siguientes al primer caso.
Se desconoce con exactitud qué causa la vasculitis. Esta inflamación puede provocar un estrechamiento de las paredes de los vasos sanguíneos que dificulta el paso de la sangre; puede cerrar por completo los vasos sanguíneos impidiendo el paso de la sangre o estirarse y debilitarse hasta que brotan y pueden llegar a reventar produciendo una peligrosa hemorragia interna.

Enfermedad de Kawasaki. Fuente: CDC
Otras Vasculitis
Existen diferentes tipos de vasculitis, entre ellas:
- Poliarteritis nudosa (PAN)
- Poliangeítis microscópica (PAM)
- Vasculitis granulomatosa y alérgica de Churg-Strauss
- Vasculitis por hipersensibilidad o vasculitis cutánea
- Granulomatosis de Wegener (GPA)
- Arteritis de Takayasu
- Enfermedad de Buerger o tromboangitis obliterante
- Enfermedad de Behçet
- Vasculitis primaria del sistema nervioso central
El tratamiento depende de la causa y, si no se identifica, se opta por un tratamiento conservador.
Comparación de PSH y EK
A continuación, se presenta una tabla comparativa con las principales características de la PSH y la EK:
| Característica | Púrpura de Schönlein-Henoch (PSH) | Enfermedad de Kawasaki (EK) |
|---|---|---|
| Afectación Principal | Pequeños vasos (IgA1) | Vasculitis aguda, puede afectar arterias coronarias |
| Edad Frecuente | 3-15 años (mayoría < 10) | 6 meses - 5 años |
| Síntomas Clave | Púrpura palpable, artritis, dolor abdominal, nefritis | Fiebre, exantema, conjuntivitis, cambios en mucosas |
| Complicaciones | Afectación renal a largo plazo | Anomalías coronarias (aneurismas) |
| Tratamiento | Soporte, corticoides (casos severos), inmunosupresores | Inmunoglobulina intravenosa, aspirina |